Advanced information
![]()
Advanced information:
Gene Modification in Mice [pdf]
Gene Modification in Mice
Introduction
The 2007 Nobel Prize in physiology or medicine is awarded to Drs Mario R. Capecchi, Martin J. Evans and Oliver Smithies for their discoveries of principles for introducing specific gene modifications in mice by the use of embryonic stem cells. Their work has made it possible to modify specific genes in the germline of mammals and to raise offspring that carry and express the modified gene. The toolbox of experimental genetic methods developed by Capecchi, Evans and Smithies, commonly called the knockout technology, has permitted scientists to determine the role of specific genes in development, physiology, and pathology. It has revolutionized life science and plays a key role in the development of medical therapy.
The discoveries
Martin Evans identified and isolated the embryonic stem cell of the early embryo, the cell from which all cells of the adult organism are derived. He established it in cell culture, modified it genetically, and reintroduced it into foster mothers in order to generate a genetically modified offspring. Mario Capecchi and Oliver Smithies, independently of each other, discovered how homologous recombination between segments of DNA molecules can be used to target genes in the mammalian genome and developed methods to generate genetically modified mice. Such animals have become indispensable in medical research. Furthermore, the knowledge concerning stem cell biology and gene technology obtained during the research that led to the “knockout mouse” has changed our understanding of normal development and disease processes and identified new avenues for medical therapy. Fig. 1 shows the general strategy for gene targeting in mice.
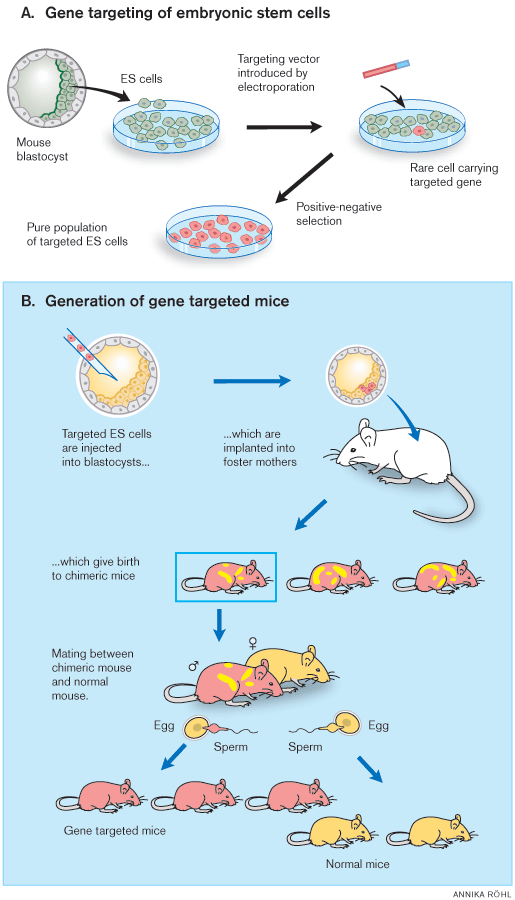 |
| Figure 1. General strategy for gene targeting in mice. A) Gene targeting of embryonic stem (ES) cells in culture is followed by cloning of an ES cell line containing the desired mutations. Positive-negative selection is used to enrich for ES cells containing the modified genes. B) These ES cells are injected into blastocysts, which are injected into foster mothers to generate chimeric mice able to transmit the mutant gene to their progeny. To facilitate isolation of the desired progeny, the ES cells and recipient blastocysts are derived from mice with different coat colour alleles. In the figure, gene targeted ES cells and their progeny are shown in red and blastocysts in yellow. |
Research that led to gene targeting
Embryonic stem cells
A stem cell is a cell that is capable of extensive proliferation, creating more stem cells (self-renewal) as well as more differentiated cellular progeny. Somatic stem cells are necessary for renewal of the tissues of the adult organism. For instance, hematopoietic stem cells in the bone marrow differentiate into blood cells, i.e. erythrocytes, megakaryocytes/platelets, and the different types of leukocytes. While each somatic stem cell of the adult organism is committed to a certain line of differentiation, the early embryo contains stem cells that are totipotent, i.e. they give rise to all cell types in the developing organism. Therefore, the thought that embryonic stem cells from the blastocyst could be used to create to a living mammalian organism has fascinated scientists for many years.
The concept that differentiated cells and tissues are derived from undifferentiated stem cells (“Stammzellen”) was already proposed a hundred years ago [1]. However, their precise properties remained elusive for many decades. Studies of testicular teratomas showed that these tumours contain totipotent cells. In the 1950s, Leroy Stevens at the Jackson Laboratory found that mice of the 129Sv strain have a high frequency of such tumours. He showed that their cells could develop into embryoid bodies, i.e. aggregates of embryonic cells. When transplanted, such aggregates could induce solid tumours with many different cell types [2, 3]. A few years later, Kleinsmith and Pierce demonstrated that such tumours were derived from undifferentiated embryonal carcinoma cells [4].
The development of cell culture techniques permitted investigators to establish cultures of embryonal carcinoma cells (EC cells) from murine testicular teratocarcinomas. Several scientists including Martin Evans at the University of Cambridge reported on such cultures in the early 70s [5-7].
Evans obtained 129Sv mice from Stevens, established a colony of mice, and characterized the teratoma derived cells in culture [8, 9]. These embryonal carcinoma (EC) cells could be grown on feeder layers of irradiated fibroblasts. When the latter were withdrawn, extensive in vitro differentiation occurred. It proceeded through a primitive embryonic endoderm, which clumped into embryoid bodies. Attachment on a solid surface gave rise to all kinds of cell types, including skin, nerve, beating cardiac muscle, etc. This showed that the EC cells differentiated in the same way as the inner cell mass of the mouse embryo [8, 9].
Evans saw the potential in using these EC cells not only for cell culture studies but also for creating chimeric mice. In order to realise this vision, he established a collaboration with Richard Gardner in Oxford, who made injections of EC cells into blastocysts and reimplanted them into foster mice. The offspring was chimeric, with contributions from EC cells in nearly every tissue [10]. Similar findings were made by several other groups at about the same time, [11] [12]. However, chimeric mice carrying EC derived cells developed multiple tumours and could not contribute to the germ line due to karyotypic abnormalities.
It became obvious to Evans that an alternative strategy had to be used if one were to obtain germline transmission derived from cultured embryonic stem cells. With the use of monoclonal antibodies, he characterised cell surface macromolecules of EC cells and their normal counterparts, thus identifying molecular markers of early differentiation [13]. The results suggested that normal cells with a similar phenotype as EC cells could be found and used for experiments. In 1980, Evans teamed up with the embryologist Matt Kaufman to combine cell culture and embryo manipulation. As described by Evans in a later review [14], he had intended to use haploid embryos for cell culture but prepared some diploid ones as controls. Evans writes [14]:
“When I cultured these blastocysts as explants in tissue culture, using a medium that had been honed for optimum cloning efficiency of both mouse and human EC cells, I immediately noted an outgrowth of EC-like cells. These cells were clearly recognizeable as the sought-after pluripotential cells, and they passed every test: They formed teratomas in vivo, and they differentiated in vitro. They bore the cell surface antigens that we expected. They stained strongly positive for alkaline phosphatase, were karyotypically normal and, most importantly, made splendid chimeras.”
These cells were the embryonic stem cells (ES cells) that became critical for the success of gene targeting. Evans and Kaufman published their report on ES cells in a seminal paper in Nature in July, 1981 [15]. Gail Martin, a former co-worker of Evans, reported similar findings half a year later [16]. In their Nature paper, Evans and Kaufman pointed out the possibility of using ES cells for gene modification. They wrote [15]:
“Their [i.e. ES cells] use as a vehicle for the transfer into the mouse genome of mutant alleles, either selected in cell culture or inserted into the cells via transformation with specific DNA fragments, has been presented as an attractive proposition. In many of these studies the use of pluripotential cells directly isolated from the embryos under study should have great advantages.”
Evans’ team set up blastocyst injection techniques to test whether indeed ES cells could contribute to functional germ cells and thus be used to create a chimeric mouse. They reported successful germline transmission in 1984, in another landmark paper in Nature [17].
The next step was to determine whether ES cells could be used to introduce genetic material into the germline. Evans and his co-workers infected ES cells with a recombinant retrovirus before injecting them into blastocysts [18]. Retroviral DNA was identified in the founders and transmitted to the F1 offspring, demonstrating introduction of the foreign DNA into the mouse germline [19]. In October, 1986, Evans et al. reported their findings in Nature and concluded that “cultured embryonic cells provide an efficient means for the production of transgenic animals” [19]. In December of that year, another laboratory reported germline transmission of a neomycin resistance gene that they had introduced into ES cells by retroviral infection [20].
Evans now took the important step of introducing a mutant form of a specific, endogenous gene into the mouse genome. He and his co-workers transferred a mutant gene for hypoxanthine phosphoribosyltransferase (HPRT), which is defective in Lesch-Nyhan syndrome, an X-linked monogenic defect of purine metabolism [21]. Several copies of the mutated HPRT gene were introduced into the genome of the ES cells by retroviral infection in culture. Mutated ES cells were injected into blastocysts and contributed to chimeras. The mutations were transmitted germline and identified in the male offspring as loss of HPRT activity. In a paper published in Nature back-to-back with the one from Evans’ lab, Hooper et al in Edinburgh reported germline transmission of another mutated HPRT gene, a spontaneous deletion mutation in ES cells [22]. For the first time, models of human disease had been created by genetic manipulation of ES cells.
In their paper [21], Evans and his co-workers point out that their success “opens up the possibility of deriving strains carrying specifically induced alterations in other genes” and suggest that “it may also eventually be possible to produce specific alterations in endogenous genes through homologous recombination with cloned copies modified in vitro”, citing the work of Capecchi and Smithies [23, 24]. Indeed, the combination of the two technologies revolutionized experimental medicine, as we now know nearly 20 years later.
Gene targeting and transgenesis in mammals
Transgenic mice
The mouse has been a favourite animal for genetic studies for many decades and was an obvious choice for the first attempts to introduce new genes into the mammalian genome. Work in several laboratories had defined conditions for manipulating fertilized mouse eggs and blastocysts in culture. Using these culture techniques, SV40 virus DNA was introduced into blastocysts, which were subsequently implanted into pseudopregnant foster mothers. SV40 DNA could be detected in the offspring but it was impossible to demonstrate with certainty whether the DNA was integrated into the host genome, or remained as episomes [25]. A few years later, the first transgenic mouse was created by infecting embryos with Moloney leukemia virus [26]. A DNA copy of the viral RNA was present in the genome of the transgenic mice and was transferred to the offspring in a Mendelian fashion, therefore virus DNA had been introduced into the mouse germline. Subsequent development has made it possible to introduce and overexpress a large number of transgenes in mice and also other mammals [27]. However, integration of the foreign DNA in the genome occurs at random and the number of copies varies. Although an important tool in life science, transgene technology of this kind lacks precision with regard to the inserted gene and cannot be used to manipulate endogenous genes in a predetermined manner. These inherent problems with the transgenic overexpression technique limit its usefulness.
Homologous recombination
The principle of recombination between homologous genes has been known for half a century and was recognized by a Nobel Prize to Joshua Lederberg in 1958 for his studies in bacteria. In the 70s, it became evident that eukaryotes employ a similar machinery to mediate exchange of genetic information between homologous chromosomes during meiosis. Early studies in yeast were followed by experiments demonstrating recombination between retroviral DNA sequences in the mammalian genome and introduced oligomeric retroviral DNA. Pioneering work by Richard Axel (2004 Nobel Prize for the discovery of odorant receptors) showed that cultured mammalian cells defective in thymidine kinase could be rescued by introduction of the herpes virus thymidine kinase (tk) gene [28]. Mario Capecchi decided to improve the method and used a fine glass pipette to inject DNA directly into the nucleus [29]. This improved the efficiency of gene transfer dramatically and Capecchi’s method was rapidly adopted by other investigators to introduce new genes into fertilized mouse embryos and produce transgenic mice [30]. However, the transferred gene was still introduced at random in the host genome.
Capecchi now made a crucial observation: when the tk gene was injected, copies were integrated in only one or two loci of the host genome, with multiple copies forming head-to-tail concatemers. He reasoned that such concatemers could only be generated by two mechanisms: either by replication or by homologous recombination. A series of careful experiments were performed, which unequivocally demonstrated that head-to-tail concatemers were generated by homologous recombination [31]. This, in turn, provided evidence that mammalian somatic cells possess an efficient enzymatic machinery for mediating homologous recombination. If this machinery could be harnessed to accomplish homologous recombination between a newly introduced DNA molecule and the same DNA sequence in the recipient cell’s genome, any cellular gene could be mutated.
Capecchi now submitted a grant proposal to the U.S. National Institutes of Health to test the feasilibity of gene targeting in mammalian cells. It was rejected since the reviewers considered it extremely unlikely that the introduced DNA would find its matching sequence within the host genome (cited by Capecchi in a later review [32])! At about the same time, Martin Evans et al in England proposed a similar strategy in a grant application to the UK Medical Research Council, which was also turned down for being over-ambitious!
Capecchi decided to continue working on homologous recombination in spite of being turned down by NIH. He generated recipient cell lines that carried a defective neomycin resistance gene (neor) and was able to repair it by introducing a functional neor gene [23]. Correction occurred at a relatively high frequency (in one cell per 1,000 injected cells), making it likely that homologous recombination could be used to manipulate genes of the mammalian genome.
In parallel with Capecchi’s work, Oliver Smithies had developed the concept that homologous recombination might be used to repair mutated genes. As early as the 1960s he had already established that an allelic variant of haptoglobin had occurred through recombinatorial events [33]. Later on, he cloned human fetal globin genes and concluded that Gγ and Aγ had arisen through a process involving homologous recombination [34]. He devised a stepwise selection procedure for recovering targeted cells carrying modified genes. The strategy was successful and he reported in a landmark paper in the September 19, 1985 issue of Nature the successful integration by homologous recombination of a plasmid into the chromosomal β-globin gene of human erythroleukaemia cells [24].
By 1985, Capecchi had shown that homologous recombination occcurs with high frequency in mammalian cells and Smithies had used homologous recombination to insert a plasmid DNA sequence into a chromosomal gene of a human cell. However, all this work was carried out in cell culture. Could homologous recombination be used to target genes in the germline and obtain strains of genetically modified animals? Both Capecchi and Smithies had heard of Martin Evans’ ES cells and decided to give them a try. With the help of Evans, they both set up ES cell culture for use in homologous recombination experiments.
Smithies first used homologous recombination to correct a mutant HPRT gene in cultured ES cells [35]. For this purpose, an ES cell line was used that carried a deletion mutation; this cell line had previously been used for production of mutant mice. The HPRT gene was repaired with a plasmid carrying the missing promoter and first 2 exons and Smithies showed that treated cells survived and grew in HAT selection medium, which requires HPRT enzyme activity. Smithies and his co-authors concluded that “This modification of a chosen gene in pluripotent ES cells demonstrates the feasibility of this route to manipulate mammalian genomes in predetermined ways” [35].
Capecchi’s team also chose the HPRT gene for their early studies. Standard methods were available for selectively growing cells with functional HPRT enzymes and had already been used for several years for selection of mutants, hybridoma cells in monoclonal antibody production etc. Thomas and Capecchi [36] introduced a neomycin resistance gene into an exon of the HPRT gene in ES cells and showed that clones of transfected cells had lost HPRT but gained neoR activity. They concluded in their Cell paper that “It is hoped that this combination of using ES cells as the recipient cell line and site-specific mutagenesis achieved by gene targeting will provide the means for generating mice of any desired genotype.” [36] They continued by outlining an experimental strategy:
“An advantage of this scenario is that the first generation chimera will usually be heterozygous for the targeted mutation and that subsequent breeding can be used to generate the homozygous animal. Thus, only one of the two loci need be inactivated, and recessive lethals can be maintained as heterozygotes. If successful, this technology will be used in the future to dissect the developmental pathway of the mouse as well as to generate mouse models for human disease.” [36]
This vision has become reality and is now a cornerstone of experimental medicine.
It was important to proceed from the “model gene” HPRT to a general strategy that would allow targeting of genes whose function cannot be selected for in cell culture. Thomas and Capecchi [36] had pointed out that the frequency of homologous recombination vs random integration was 1/1,000, which should be high enough to permit targeting of non-selectable genes as well. This observation prompted work to develop the methods needed for such approaches. The following year, Capecchi’s positive-negative selection strategy for enriching ES cells containing a targeted disruption of any transfected gene was presented in Nature [37] (Fig 2). A neomycin resistance element (neoR) is introduced into an exon of the replacement vector, which also has a thymidine kinase (HSV-tk) element at its end. Homologous recombination of the targeted gene will result in neoR expression but the tk element will be lost since it was outside of the recombining DNA sequences. In contrast, random integration of the replacement vector will introduce tk as well as neoR into the gene. This strategy was successfully used to disrupt the int-2 gene, which is a member of the fibroblast growth factor (FGF) family [37].
All the components were now in place for producing gene-targeted mouse strains: the development of ES cell culture, the demonstration that gene modification in such cells can be transmitted to the germline and registered in the offspring, the observation that homologous recombination occurs with high frequency in the mammalian genome, the application of gene transfer methods to ES cells, and the invention of strategies for enriching transfected cells. Several laboratories joined the race and 1989 saw the birth of several different knockout mice [38-41]. Fig 3 shows the molecular evidence for correction of the mutant HPRT gene in the germline by Smithies and co-workers [39].
Further development of gene targeting technology
After the establishment of gene targeting technology, several important modifications and developments, in several laboratories, have extended its use in significant ways. An ingenious development of gene targeting has been made by introducing recognition sites for the enzyme Cre recombinase, so-called loxP sites, into existing genes. When mice carrying such “floxed” genes are mated with transgenic mice expressing Cre recombinase, the target gene of the offspring is modified through Cre action [42-44]. Another site-specific recombinase, Flp, is also frequently used to construct conditional targeting of genes in mice [45]. The activity of the Cre, or Flp, gene can be controlled by placing it under a suitable promoter to achieve tissue-specific gene targeting [46]. Expression of Cre and hence targeting of the floxed gene can be restricted to e.g. T cells (lck promoter), cardiac muscle (cardiac myosin promoter), neurons (enolase promoter) or epithelia (cytokeratin promoter).
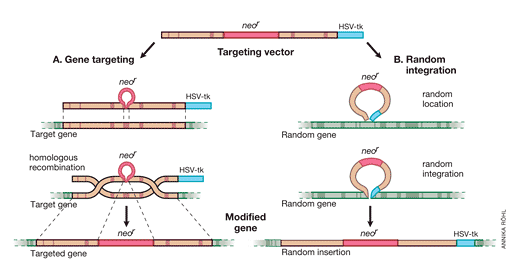
Fig. 2. Positive-negative selection is used to enrich for ES cells containing a targeted disruption of a gene. In both gene targeting (A) and random integration (B), the upper line shows the targeting vector, the middle one the chromosomal gene, and the lower one the modified gene. Genes targeted by homologous recombination contain the neoR element but not HSV-tk, since the latter resides outside the sequences in the targeting vector homologous to sequences in the chromosomal gene. In contrast, random integration of the vector results in introduction of HSV-tk as well as neoR. Screening methods select positively for neoR positive clones and discard those neoR + clones that carry HSV-tk.
Cre expression can also be controlled temporally, by introducing an element into the promoter which requires a ligand such as a drug for induction [47]. Tetracyclin, type I-interferon and tamoxifen (which binds to an estrogen receptor-binding element) have all been used to obtain drug-inducible promoters. In this way, a desired gene can be targeted by administrating the drug. By introducing a tamoxifen site into a tissue-specific promoter, gene targeting can be obtained selectively in a certain tissue when the mouse is treated with the drug.
Cre-lox technology can also be used to replace an existing gene with another one [48]. Such “knock-in” has been used e.g. to replace murine immunoglobulin or MHC genes with human ones in order to “humanize” the mouse with regard to immune function. It has also been used to replace an allele with another one, the latter for instance being an allele suspected to cause disease.
Significance of gene targeting for physiology and medicine
Gene targeting has transformed scientific medicine by permitting experimental testing of hypotheses regarding the function of specific genes. Prior to gene targeting, our understanding of the role of genes in higher organisms was deduced from observations of spontaneous mutations in patients and experimental animals, linkage and association studies, administration of gene products to animals and, to some extent, from cell culture experiments. However, cell culture is not helpful for understanding functions and diseases involving multicellular, integrative responses. Insights into organ systems such as the nervous system,the cardiovascular system, and the immune system, were fragmentary at best, as was knowledge of mammalian development. As the cardiovascular physiologist Heimo Ehmke put it, “cells don’t have blood pressure” [49]. The possibility of observing the effects on the intact organism of destroying a candidate gene transformed these areas of research. For instance, cardiovascular physiologists switched from rats to mice as models, downscaling their instruments and techniques in order to study the genetic regulation of hemodynamics. A new era of genetic physiology was born.
The genomes of man and mouse contain about 22,400 genes. Several thousand of them have already been investigated by gene targeting. Collectively, these studies have provided a wealth of information about gene function in development and disease. They have helped fuse mechanistic molecular biology with integrative life sciences such as embryology, physiology and immunology and have prompted new technical developments in physiological sciences. For medicine, the modeling of human diseases by gene targeting in mice has been particularly informative.
At this stage, it may be helpful to recapitulate the criteria first proposed by Claude Bernard for the scientific method in medicine [50]: Medical scientists use observations, hypotheses and deductions to propose explanations, theories, for natural phenomena. Predictions from these theories are tested by experiment. Any theory which is cogent enough to make predictions can be tested reproducibly in this way. Therefore, the scientific method is essentially a cautious means of building a supportable, evidence-based understandingof our natural world. Experiments are crucial in this process.
Prior to gene targeting, genetic medicine lacked the means for experimental testing. If we make an analogy with Robert Koch‘s approach to infectious diseases [51], genetic medicine could apply the first of Koch’s postulates (i.e. observe an association between microbe, or in this case, gene or allele, and disease) and with the advent of gene cloning, the second one (isolate the microbe/gene from the diseased individual and establish it in culture), but applying Koch’s third postulate (induce the disease by transferring the microbe/gene to a host organism) required gene targeting. By mutating a gene to destroy its function (knock-out) or switching it to a disease-associated allele (knock-in), disease is induced if the hypothesis is correct. Alternative approaches based on genetic epidemiology are currently being developed but currently available methods do not have the precision of hypothesis-based experiments. This digression into scientific theory may suffice to make the point that only by targeting candidate genes did it become possible to formally establish causality between gene and disease. Let us now look at some specific examples of the impact of gene targeting in medicine.
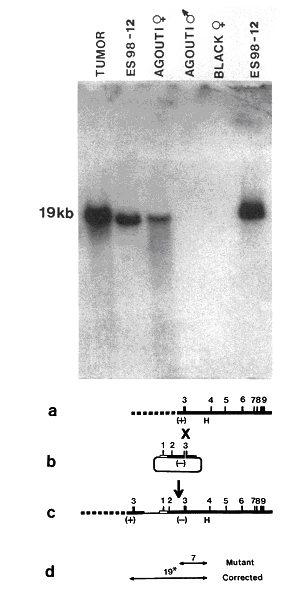 |
Figure 3. Molecular genetic evidence for germline transmission of repaired HPRT gene. From ref [39]. The Southern blot shows genomic DNA hybridized to a probe specific for a sequence in the targeting vector. ES cells were from agouti mice, blastocysts from black mice and the HPRT gene resides on the X chromosome. The tumor was from a chimeric mouse that carried the targeted gene, as did the targeted ES cell line. In the F1 offspring, female agouti mice derived from targeted ES cells carried the targeted HPRT gene, while it was present neither in black mice derived from recipient blastocysts nor in male agouti mice. The lower part shows the targeting strategy, with the deletion mutation in the HPRT gene (a), the targeting construct (b), the HPRT gene corrected by gene targeting (c), and the probe (19*) used for hybridization (d). Reprinted, with permission, from Proceedings of the National Academy of Sciences. Copyright 1989 National Academy of Sciences, U.S.A. |
Monogenic diseases
The first area to which experimental geneticists turned their attention after the birth of gene targeting in mammals was monogenic diseases. The Lesch-Nyhan syndrome, a defective nucleotide metabolism caused by a mutation in the HPRT gene, actually served as the model condition during development of the technology, both in Evans’ and Smithies’ laboratories (see above). One of the reasons for chosing this particular medical condition was because selection conditions for isolating transduced cells were available for HPRT. The first examination of HPRT-/- mice was disappointing since neither neuropathological nor behavioural features of human disease could be observed [21, 39]. This prompted analysis of purine salvage pathways in mice and led to the findings that mice depend largely on adenine phosphoribosyltransferase (APRT) for purine salvage and are therefore not as sensitive to HPRT deficiency as humans. Administration of an APRT inhibitor to HPRT-/- mice induced persistent self-injurious behaviour resembling the clinical features in human disease [52]. This is an illustration of the need for sophisticated analysis of integrative functions when characterising the phenotype of gene-targeted mice.
Cystic fibrosis is one of the most common monogenic disease and was chosen for gene-targeting studies by Smithies and his co-workers [53, 54]. The defective gene had been identified by linkage studies in patient families followed by molecular cloning. It turned out to be a cAMP-activated choride channel and was termed cystic fibrosis transmembrane conductance regulator (CFTR). By knocking out CFTR in mice, a condition was generated that reproduced many features of the human disease. Thus, CFTR-/- homozygotes displayed defective chloride transport in epithelia of airways and intestines, failure to thrive, meconium ileus, and pathological alterations of gastrointestinal glands. These studies were among the first to create a model of a human disease by gene targeting in mice. They have been followed by an avalanche of such knock-out models.
The pathogenesis of inherited heart diseaseshave been explored successfully by gene targeting approaches [55, 56]. For instance, targeting of genes encoding components of the contractile apparatus in cardiomyocytes leads to cardiomyopathy; targeted mutations in connexin proteins of gap junctions cause conduction defects; disrupted genes for transcription factors involved in heart development lead to congenital heart malformations; and targeting of genes controlling energy metabolism causes cardiomyopathy.
Complex diseases
Complex diseases involving the action of more than one gene, and in addition, gene-environment interactions, represent a particular challenge for medical research. Inheritance, penetration and interactions are usually poorly understood, it has been difficult to dissect the contribution of an individual genetic factor, and the distinction between causation and correlation has been problematic. In order to prove causation in such a complex system, experiments must permit detection of the effects of changing only a single variable at one time. Gene targeting made such experiments possible and has permitted proof of causation in complex diseases.
Cardiovascular diseases
Oliver Smithies has been the leader in this development. Together with Nobuyo Maeda, he focused on two important, complex diseases, hypertension and atherosclerosis (reviewed in [57]). Twin studies suggest that genetic factors may account for approx 70% of familial aggregation of essential hypertension. However, at least 10 genes have been shown to alter blood pressure and their gene products appear to interact in complex ways. In spite of the discovery that angiotensinogen (AGT) gene polymorphism is associated with essential hypertension, the genetics of this disease has remained poorly understood [58]. Little is known about the number of genes actually involved in human essential hypertension, their quantitative effect on blood pressure, their mode of transmission, or their interaction with other genes and environmental components.
Smithies suspected that gene dose effects would impact on blood pressure levels and designed a new method for titrating gene dosage by producing mice with one, two or three functional copies of the AGT gene [59]. “Conventional” gene targeting was used to produce the one- and two-copy mice and gap-repair gene targeting to produce mice with three copies of the AGT gene. This resulted in proportionally higher levels of gene products (i.e. plasma angiotensinogen protein) and, importantly, proportionally higher blood pressure with increasing gene copy number. When Smithies et al targeted another important gene for blood pressure regulation, the one coding for the angiotensin-converting enzyme (ACE), no such linear relationship was observed, in spite of the effectiveness of ACE inhibitors in reducing blood pressure. The investigators submitted their data to a computer simulation for complex interacting systems and could propose a model for blood pressure control through the renin-angiotensin system, which has proven to be useful for understanding essential hypertension [60]. It shows that gene dosage, gene expression, and gene product clearance/catabolism must all be considered when evaluating the genetic regulation of blood pressure.
In 1992, Nobuyo Maeda, working in Smithies’ department at the University of North Carolina, developed a mouse model of atherosclerosis by targeting the gene for apolipoprotein E (Apoe) [61]. The same gene was targeted independently by investigators at Rockefeller University [62]. The Apoe-/- mouse develops spontaneous atherosclerosis which is remarkably similar to human disease. The following year, Michael Brown and Joseph Goldstein (1985 Nobel Prize for discoveries concerning cholesterol metabolism) and their co-workers targeted the gene for the low density lipoprotein (LDL) receptor (Ldlr) and obtained a mouse that develops atherosclerosis when fed a cholesterol-rich diet [63]. The introduction of the two mouse models with defective Apoe and Ldlr genes have completely changed atherosclerosis research. By crossbreeding them with other gene-targeted mice, it has been possible to deduce the importance of genes regulating inflammation, lipid metabolism, blood pressure and other factors proposed to be involved in atherosclerotic cardiovascular disease [64]. They are also used abundantly in the pharmaceutical industry for development and testing of new drugs against coronary artery disease.
Cancer
Gene targeting has been exceptionally useful in cancer research. A large number of protooncogenes, tumor suppressor genes, angiogenetic factors etc have been targeted in different tissues in mice to shed light on the induction and spreading of tumours [65]. Gene targeting of tumour suppressor genes have helped clarify their role in the formation of tumours. For instance, mice carrying a targeted p53 gene were predisposed to tumour development [66]. Conditional targeting (using Cre-lox technology) of the adenomatous polyposis coli (APC) gene induces colorectal tumors in mice and APC-targeted mice have become useful models for research on solid tumours [67]. Targeting of genes for endothelial growth factors and proteolytic enzymes have been essential for understanding mechanisms of neoangiogenesis and metastasis of solid tumours and are also used for developing therapeutic strategies to prevent spreading [68].
Other diseases
Contemporary research into most if not all major human diseases involves gene targeting in mice and there are “knockout models” for endocrine, metabolic, neurological, inflammatory and other disorders. Gene-targeted mouse models have also become increasingly important in studies of host defense against pathogens. Indeed, gene targeted mice have become indispensable in virtually all aspects of medical research.
Conclusions
Gene targeting has transformed physiology and medicine. Among the basic biomedical sciences, it is difficult to imagine contemporary medical research without the use of gene targeted models. The ability to generate predictable designer mutations in mouse genes has led to penetrating new insights into development, immunology, neurobiology, physiology, and metabolism. It has also allowed disease models of human pathologies to be generated in a tractable mammalian system and consequently enabled experimental dissection of disease states, identification of new therapy targets and the development of test systems for pharmacology. Finally, it is obvious that the development, in the future, of novel therapies to correct genetic defects in man will build on the experience of gene modification in mice that is based on the discoveries made by Mario Capecchi, Martin Evans and Oliver Smithies.
Göran K Hansson
Professor of cardiovascular research at Karolinska Institutet
Member of the Nobel Committee for Physiology or Medicine
Nobel Prizes and laureates
Six prizes were awarded for achievements that have conferred the greatest benefit to humankind. The 14 laureates' work and discoveries range from quantum tunnelling to promoting democratic rights.
See them all presented here.

